
L’amyotrophie spinale SMA
Extrait du site myonet.org
1.Une maladie neuromusculaire
En temps normal, les muscles de notre corps se contractent sous l'impulsion d'un signal provenant du cerveau. Ce signal est transmis par l'intermédiaire des neurones de la corne antérieure de la moelle épinière (aussi appelés motoneurones inférieurs). Chaque muscle est relié à la moelle épinière par un nerf périphérique. On appelle unité motrice l'ensemble constitué d'un motoneurone périphérique et les fibres musculaires qui lui sont reliées. La transmission de l'influx nerveux entre le motoneurone et la fibre musculaire se fait par l'intermédiaire de la jonction neuromusculaire grâce à un composé chimique.
L’amyotrophie spinale est une maladie génétique est due à l’altération (mutation) ou à l’absence (délétion) du gène nommé SMN1. Ce gène défectueux ou absent n’est pas capable de donner les bonnes informations pour produire une protéine appelée SMN (protéine de "survie du motoneurone"). Lorsque la protéine SMN n'est pas produite en quantité suffisante, le bon fonctionnement des motoneurones, ou neurones moteurs, est alors impossible. Les ordres de mouvement ne sont plus transmis entre la moelle épinière et les muscles. Les motoneurones permettant de « donner des ordres » aux muscles finissent ensuite par mourir. Les muscles concernés deviennent inactifs, s’affaiblissent et s’atrophient, entraînant une dégradation des articulations, des ligaments et des os. La maladie conduit ainsi à des problèmes respiratoires sévères ainsi qu'à des déformations physiques irréversibles.
L’évolution des amyotrophies spinales est variable : généralement, plus le début est précoce (dès la naissance ou au cours des premiers mois de la vie, gênant le développement de l’enfant), plus le pronostic est sévère.
Cette maladie touche tous les muscles des membres inférieurs et supérieurs, notamment ceux utilisés pour la marche, pour le contrôle des bras, des mains, de la tête et du cou, pour la déglutition ou encore pour la respiration. Cette maladie n'affecte aucunement les capacités intellectuelles. Il n’existe pour l’instant ni moyen de guérison ni traitement.
- La SMA peut toucher n’importe qui, indifféremment de l’âge, de la race ou du sexe.
- Une personne sur 40 est porteur "sain" de la maladie.
- La maladie peut se déclarer à n'importe quel âge, du nourrisson jusqu'à l'âge adulte.
- Un enfant sur 6 000 à 10 000 naît avec la maladie.
- La SMA est la première cause de décès d’origine génétique des enfants de moins de 2 ans.
2.Une maladie génétique
Chaque partie du corps est composée de cellules. Chaque cellule possède un matériel génétique qui détermine comment cette cellule doit fonctionner. Les unités de base de ce matériel génétique sont appelées gènes. Les gènes ne peuvent pas être vus individuellement au microscope, aussi puissant soit-il, mais ils sont localisés sur des structures linéaires appelées chromosomes.
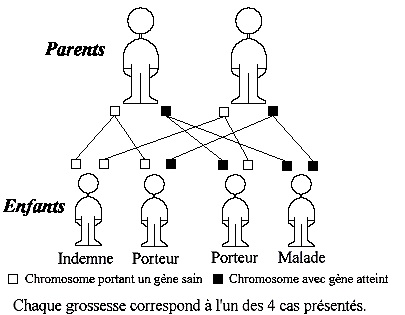
Chaque personne possède deux copies de chaque gène, chacune provenant d'un de ses parents.
Dans le cas d'une maladie récessive, il faut que les deux copies du gène responsable de la maladie soient affectées pour qu'une personne soit atteinte de la maladie. Ainsi, les personnes qui possèdent un seul gène affecté (absent, erroné ou incomplet) et un gène normal, ne seront pas atteintes mais elles seront porteuses de la maladie.
En conséquence, le diagnostic d'une maladie récessive montre que les parents sont porteurs de la maladie (en général), et qu'ils ont pour chaque grossesse une probabilité de 25 % d'avoir un enfant atteint de la maladie, de 50 % d'avoir un enfant porteur qui ne développera pas la maladie, et de 25 % d'avoir un enfant totalement exempt de gêne affecté. Il existe parfois une mutation spontanée d'un des gènes, l'autre gène affecté étant hérité d'un parent, ce qui provoque l'apparition de la maladie alors que les deux parents ne sont pas tous les deux porteurs. Ce cas de figure est plutôt exceptionnel (environ 2% des cas d'amyotrophie spinale). Le terme "autosomique" signifie qu'une maladie génétique n'est pas liée aux gènes caractérisant le sexe (X et Y). Elle touche donc indifféremment les filles et les garçons. L'amyotrophie spinale est une maladie autosomique récessive.
3.Les principales formes
Il existe trois formes d’amyotrophie spinale infantile et une forme adulte, qui sont répertoriées selon l’âge d’apparition des premiers symptômes et la rapidité de leur évolution. Il faut cependant relativiser cette classification, l'évolution de la maladie et la gravité de son atteinte étant très variables suivant les individus.
La forme de type I, la plus grave, est aussi connue sous la désignation d’amyotrophie spinale infantile sévère ou maladie de Werdnig-Hoffmann. Elle apparaît pendant la vie intra-utérine ou au cours des premiers mois de la vie, et se traduit par une faiblesse musculaire prononcée. L’espérance de vie des enfants atteints dépasse rarement trois ans et se limite souvent à quelques mois.
La forme de type II, aussi appelée amyotrophie spinale intermédiaire (ASI), apparaît habituellement entre l’âge de six mois et de trois ans. Bien qu’elle ne soit pas aussi grave que la forme de type I, de nombreux enfants qui en sont atteints ne parviennent jamais à s’asseoir sans soutien et rares sont ceux qui parviennent à marcher ou à ramper. De fréquentes infections respiratoires peuvent compliquer la cause de cette affection et diminuer l’espérance de vie.
La forme de type III, aussi appelée amyotrophie spinale juvénile ou maladie de Kugelberg-Welander, est moins invalidante. Elle se manifeste plus tard et n’influe pas sur l’espérance de vie (qui peut varier en fonction du degré d’atteinte respiratoire).
La forme de type IV, ou amyotrophie spinale adulte, apparaît plus tard dans la vie, habituellement entre 15 et 50 ans. Elle occasionne un degré d’invalidité souvent moins prononcé et n’influe généralement pas sur l’espérance de vie. Elle est moins répandue et moins bien comprise que les trois formes infantiles.
4.Diagnostic
Dans 95 % des cas, le diagnostic est possible à partir d’une simple prise de sang. On fait une analyse de l’ADN. Cette analyse permet de mettre en évidence l’absence du gène SMN. Cela permet également d’éviter la biopsie musculaire. Les parents porteurs voulant avoir d’autres enfants, peuvent bénéficier du diagnostic prénatal, il s’agit soit :
- d’une ponction de trophoblaste effectuée à 10 semaines d’aménorrhée,
- d’une ponction du liquide amiotique effectuée entre le 3ème et le 4ème mois de grossesse.
L'amyotrophie spinale de type 2 (ASA de type 2) est une forme infantile chronique d'amyotrophie spinale proximale caractérisée par une faiblesse musculaire et une hypotonie dues à la dégénérescence et la perte des motoneurones antérieurs de la moelle épinière et des noyaux du tronc cérébral.
Sa prévalence estimée est d'environ 1/70 000. La maladie est un peu plus fréquente chez les garçons que chez les filles.
Elle débute entre 6 et 18 mois (vers 15 mois). Les enfants arrivent difficilement à s'asseoir seuls et n'arrivent pas à se mettre debout ni à marcher à un an. La faiblesse musculaire (presque toujours symétrique) touche avec prédilection les muscles des jambes et du tronc. Un tremblement des doigts est fréquent. Une insuffisance respiratoire, une scoliose, et des fractures spontanées par traumatisme minime sont fréquents.
Comme les autres formes d'ASA, l'ASA de type 2 est surtout due à des délétions homozygotes du gène SMN1 codant la protéine de survie du motoneurone (SMN). Bien qu'il ait des variations, la sévérité de l'ASA est inversement proportionnelle au nombre de copies du second gène, SMN2 et les patients atteints d'ASA de type 2 possèdent en moyenne trois copies du gène SMN2. Des délétions du gène NAIP ont aussi identifiées et pourraient influencer la sévérité de la maladie.
Le diagnostic repose sur les antécédents et l'examen clinique et il peut être confirmé par l'examen génétique. Un examen électromyographique et une biopsie musculaire peuvent être nécessaires.
La transmission est autosomique récessive mais 2% des cas environ sont dus à des mutations de novo.
Des études cliniques sont en cours pour identifier des médicaments potentiels de l'ASA de type 2 visant surtout à augmenter les taux de SMN complète. Cependant, actuellement, le traitement reste symptomatique, et son approche multidisciplinaire a pour but d'améliorer la qualité de vie. Une assistance respiratoire est nécessaire. Une ventilation non invasive peut être utile. La physiothérapie et l'ergothérapie sont recommandées. Une antibiothérapie est utilisée en cas d'infection pulmonaire. La scoliose peut nécessiter l'utilisation d'un corset avec minerve de contention, ou une correction chirurgicale.
L'espérance de vie est variable. Avec un traitement adapté, en particulier de l'insuffisance respiratoire, la plupart des patients arrivent à l'âge adulte, mais sans pouvoir marcher seuls.
5.Prise en charge et prévention
La prise en charge est naturellement fonction du degré de l’atteinte. En général, cette prise en charge s'exerce principalement de façon orthopédique et respiratoire. Une prise en charge précoce et régulière permettra de limiter, voire de prévenir certaines conséquences de la maladie. la prise en charge précoce doit permettre de limiter les infections respiratoires, les douleurs et les déformations secondaires à la fonte musculaire et contribue à assurer le meilleur développement possible de la personne atteinte. Les orthèses spécialisées, la kinésithérapie ainsi que la chirurgie orthopédique aident à préserver le mouvement, à maintenir la capacité fonctionnelle et à assurer le confort des malades.
Les éléments de prise en charges cités ci-dessous sont donnés à titre d'exemple car chaque cas d'amyotrophie spinale est unique : par exemple, certains enfants ont besoin d’une assistance respiratoire, d’autres pas. Les médecins adapteront la prise en charge en fonction de chaque cas.
- Prise en charge de la fonction respiratoire
- Prise en charge du déficit musculaire
6.Conséquences sur la vie quotidienne
Certains appareillages peuvent sembler encombrants et impressionnants, mais ils ont toujours pour objectif de soulager le malade et de faciliter son maintien et l’amplitude de ses mouvements. Une adaptation du mobilier, de l’habitation (lit, salle de bain, escaliers...) ou des véhicules peut par conséquent être utile pour que la personne atteinte puisse se déplacer de façon autonome.
Si une trachéotomie est réalisée, l’adaptation du malade et de son entourage peut nécessiter du temps. En effet, cette opération est impressionnante, puisqu’il s’agit d’une petite ouverture dans la gorge. Le malade aura notamment des difficultés à avaler (surtout les premiers jours) et à parler : l’air ne passe plus par le larynx et donc ne fait plus vibrer les cordes vocales (il sort directement par l’orifice de trachéotomie). Pour parler, il faut interrompre la sortie d’air avec un bouchon spécial et cela nécessite un apprentissage, au même titre que les règles d’hygiène à respecter.
De même, la gastrostomie est un dispositif pouvant effrayer a priori, mais il faut savoir que c’est un réel gain de confort à la fois pour l’enfant et pour les parents qui ne s’inquiètent plus à chaque repas.
Afin de permettre à l’enfant (ou à l’adulte) d’entretenir le mieux possible sa mobilité et de bouger plus facilement, des exercices en piscine (encadrés au début par un kinésithérapeute) ou en milieu aquatique sont recommandés et souvent très appréciés. Par ailleurs, il est essentiel d’avoir une hygiène alimentaire équilibrée pour maintenir une fonction musculaire optimale et n’être ni trop maigre ni en surpoids.
Il y a plusieurs moments au cours de l’évolution de l’amyotrophie spinale où la famille et le malade peuvent ressentir le besoin de trouver un soutien psychologique. Pour les parents et les grands-parents, l’annonce du diagnostic, avec la culpabilité liée au fait que l’on a transmis une maladie sans le savoir et bien entendu sans le vouloir, puis l’accompagnement de son enfant en apprenant à le soigner sans le surprotéger, sont des exemples où une aide psychologique peut s’avérer utile. Les frères et sœurs peuvent quant à eux ressentir de la culpabilité ou même de la jalousie, et une aide extérieure peut permettre de rétablir la communication au sein de la famille. A tous ces moments, la famille ne doit pas hésiter à se faire soutenir par un psychologue.
Pour les enfants ou les adultes malades, c’est le besoin d’apprendre à se prendre en charge, les difficultés à réaliser certaines activités de façon autonome, les périodes de déni ou d’opposition, comme à l’adolescence, qui sont spécialement sensibles. Pour les enfants SMA de type III, ayant appris à marcher, la perte progressive de cette capacité, lorsqu’elle survient, est particulièrement difficile à vivre. En grandissant, la confrontation au regard des autres peut être un passage délicat où un accompagnement psychologique peut être utile.
La kinésithérapie et les prises en charge orthopédique, nutritionnelle et respiratoire sont fondamentales pour éviter les complications et s’assurer que l’enfant grandisse le mieux possible. Il est très important que l’entourage et les personnes malades en soient conscients, afin de respecter au mieux les conseils d’appareillage et les exercices.
Les personnes atteintes de SMA sont suivies dans des consultations pluridisciplinaires spécialisées dans les maladies neuro-musculaires. Le suivi orthopédique de l’enfant doit être précoce et permettre d’adapter régulièrement les appareillages (corsets, coques, fauteuils) au fur et à mesure de la croissance. Des évaluations périodiques de l’état ventilatoire (respiration) du malade aident le médecin à mettre en place une assistance respiratoire à temps, si nécessaire, et à adapter régulièrement les réglages.
